不可治愈慢性肺疾病,关注罕见病系列之特发性肺纤维化
发布时间:2024-09-02
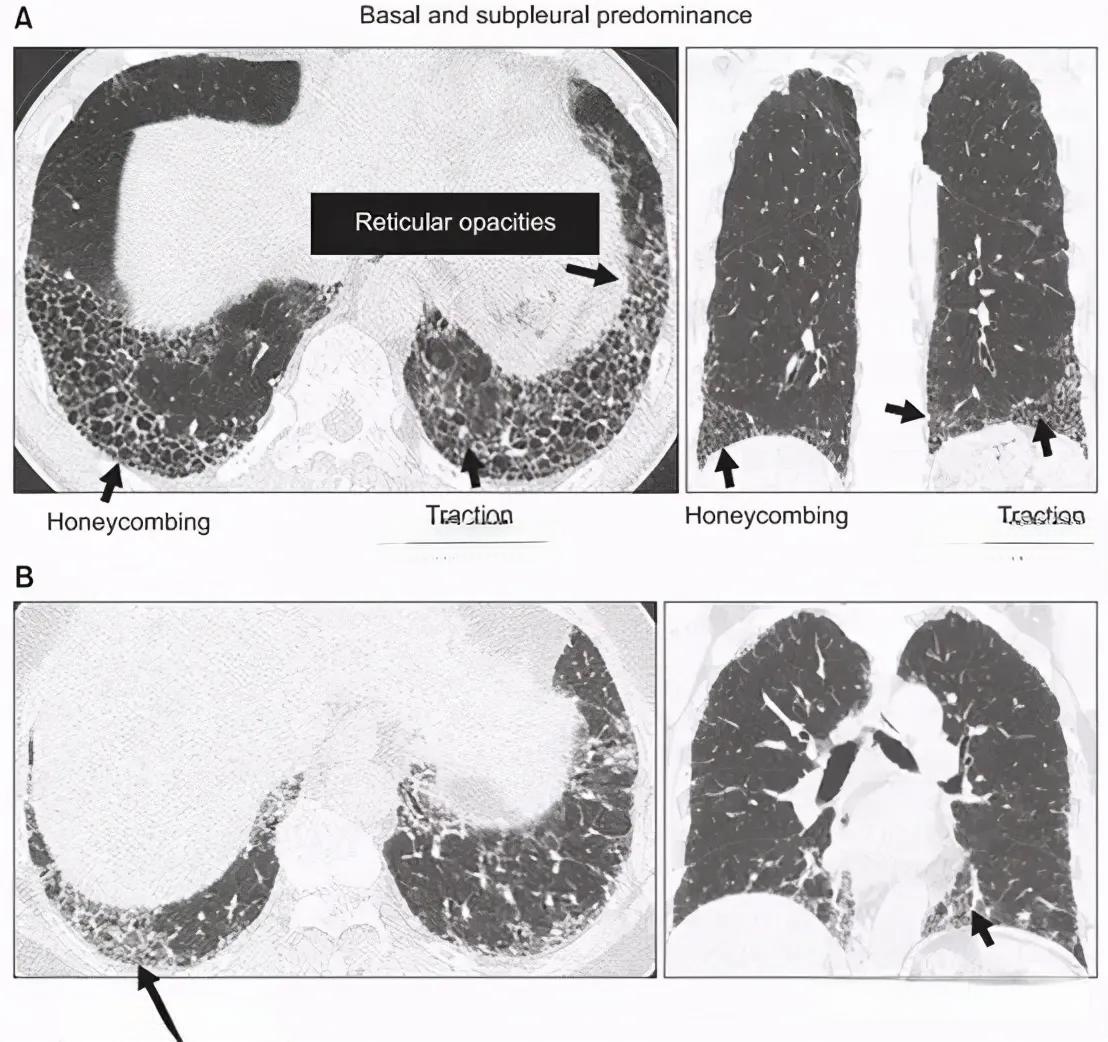
特发性肺纤维化是一种进展迅速且致命的肺部疾病,患者平均存活期仅为3-5年。长期以来,这种罕见病的发病机理不明,缺乏有效的治疗方法,成为医学界的一大挑战。然而,近期中国科学家在这一领域取得了重大突破,为患者带来了新的希望。
北京生命科学研究所/清华大学生物医学交叉研究院汤楠实验室与中日友好医院代华平团队及普沐生物科技有限公司研发团队合作,揭示了特发性肺纤维化发生的细胞和分子机制,并找到了有望治疗该病的新靶点。这一研究成果近日发表于国际学术期刊《细胞—干细胞》。
研究团队发现,肺泡再生障碍导致损伤后激活的肺泡干细胞被卡在分化的中间状态,暴露于持续升高的机械张力,这是诱发肺纤维化从肺叶边缘起始并不断向肺中心进行性发展的关键驱动因素。进一步研究发现,处于中间态的肺泡干细胞高表达多种促纤维化的基因,其中双调蛋白AREG在驱动肺纤维化发生和进展中起着关键作用。
通过一系列体外细胞实验和体内动物模型验证,研究团队证明中间态肺泡干细胞通过持续性分泌的AREG直接作用到表达EGFR的成纤维细胞上,促进了肺纤维化发生。在患者肺部样本中也得到了同样的结果。此外,研究发现特发性肺纤维化患者血清中的AREG水平显著升高,并与患者的肺功能呈显著负相关,与病情严重程度呈显著正相关。
基于这一发现,研究团队使用中和抗体有效阻断AREG,显著抑制了小鼠肺纤维化的发生和发展,提高了动物的生存率。这一结果凸显了抗AREG抗体在减缓特发性肺纤维化进展方面的治疗潜力,为开发新的治疗方法提供了重要方向。
相比之下,目前特发性肺纤维化的治疗方法有限。吡非尼酮和尼达尼布是仅有的两种获FDA批准的药物,虽然可以减缓肺功能下降,但无法逆转疾病进程。此外,肺康复治疗、氧疗和肺移植等方法也只能缓解症状或延缓疾病进展。
这一研究进展不仅深化了我们对特发性肺纤维化发病机制的理解,也为开发更有效的治疗方法提供了新的思路。抗AREG抗体作为一种潜在的治疗方法,具有靶向性强、副作用小的优势,有望为患者带来更好的治疗效果和生活质量。
然而,从基础研究到临床应用仍需时日。研究团队将继续深入探索AREG在肺纤维化中的作用机制,并开展更多的临床前研究,为未来的人体临床试验奠定基础。同时,他们还将探索AREG是否参与其他类型肺纤维化的发展,以扩大这一发现的临床应用范围。
这一研究成果不仅为特发性肺纤维化患者带来了新的希望,也展示了中国科学家在罕见病研究领域的实力和贡献。它提醒我们,即使面对最棘手的医学难题,持续的研究和创新终将带来突破。未来,随着更多类似的研究进展,我们有理由相信,特发性肺纤维化等罕见病终将不再“无药可治”。